

Clinical Trials for Ages 6 Years to <12 Years
Studied in Over 250 Patients Age 6 Through 11 Years
STUDY DESIGN
Trial 3 | Open-Label Safety Study
Trial 3 was a 24-week, open-label study (N=58) assessing the safety, tolerability, and pharmacokinetics of ORKAMBI in patients age 6 through 11 years with CF who were homozygous for the F508del-CFTR mutation, were clinically stable, and had a percent predicted FEV1 of ≥40%. Patients received ORKAMBI tablets (lumacaftor 200 mg/ivacaftor 250 mg q12h) with fat-containing food and continued to take their prescribed CF therapies (including during the 2-week washout period). Primary endpoints were safety and tolerability, including assessments of adverse events, clinical laboratory values, and spirometry (FEV1) up to 24 weeks.1,2
Trial 3 | Limitations
- The study was open-label and not placebo controlled2; therefore, causality cannot be attributed to drug effect
Additional Disclosure
- The Trial 3 efficacy results are not included in the approved full Prescribing Information, and the FDA did not consider either Trial 3 or Trial 4 in approving ORKAMBI2
SAFETY PROFILE
Primary Endpoints Were Safety and Tolerability, Including Assessments of Adverse Events, Clinical Laboratory Values, and Spirometry (FEV1) Up To 24 Weeks.

Safety Profile in Trial 3 Was Similar to That Observed in Patients Age 12 Years and Older1
Discontinuations Due to Serious Adverse Events1
- Trial 3: 3.4% (n=2); due to elevated liver transaminases (1) and rash (1)2
Serious Adverse Reactions
- Trial 3: 6.9% (n=4); included infective pulmonary exacerbation (2), ileus (1), and elevated liver transaminase levels (1)2
Liver-Related Adverse Reactions2
Potentially Clinically Significant (PCS) Laboratory Tests
| Parameter PCS/Categorical Criteria | TRIAL 3: 24 Weeks n/Na (%) |
|---|---|
| ALT or AST | ORKAMBI |
| >8 x ULN | 3/57 (5.3) |
| >5 x ULN | 5/57 (8.8) |
| >3 x ULN | 11/57 (19.3) |
| Discontinuation due to transaminase elevations | 1/57 (1.8) |
aNumber of subjects with at least one measurement during the period from initiation of study drug to 28 days following last dose.2
- In Trial 3, no patients had an increase in total bilirubin levels >2 x ULN1
Respiratory Adverse Events of Special Interest2
| TRIAL 3: 24 Weeks n/N (%) |
|
|---|---|
| ORKAMBI | |
| Total Incidence | 4/58 (6.9) |
| Wheezing | 2/58 (3.4) |
| Dyspnea | 1/58 (1.7) |
| Respiration abnormal | 1/58 (1.7) |
Common Adverse Reactions in Trial 32
Most Frequently Observed Treatment-Emergent Adverse Events in ≥10% of Patients
| TRIAL 3b: 24 Weeks | |
|---|---|
| ORKAMBI n=58 |
|
| Any TEAEs, n (%) | 55 (94.8) |
| Cough | 29 (50.0) |
| Infective pulmonary exacerbation | 12 (20.7) |
| Nasal congestion | 12 (20.7) |
| Headache | 12 (20.7) |
| Abdominal pain upper | 8 (13.8) |
| Sputum increased | 8 (13.8) |
| ALT increased | 7 (12.1) |
| Pyrexia | 6 (10.3) |
| Vomiting | 6 (10.3) |
| Abdominal pain | 6 (10.3) |
| Fatigue | 6 (10.3) |
| Nausea | 6 (10.3) |
bTrial 3 Safety Set.
OTHER RESULTS
Lung Function Through 24 Weeks
Trial 3: Percent Predicted FEV1 at Week 24 (Part of the Safety Assessment)
- +2.5% point LS mean within-group improvement2
- -3.2% point LS mean within-group decrease from Week 24 at Week 26 (washout)2
Trial 3: Sweat Chloride Was Reduced at Week 242
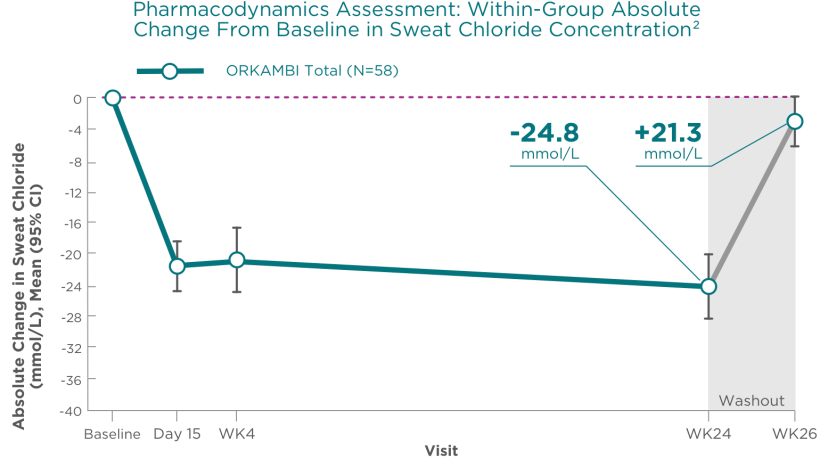

- -24.8 mmol/L LS mean within-group improvement at Week 242
- Washout: +21.3 mmol/L LS mean within-group absolute change from Week 24 at Week 262
There was no direct correlation between changes in sweat chloride levels and improvement in lung function (percent predicted FEV1)1
ALT, alanine aminotransaminase; AST, aspartate aminotransaminase; CI, confidence interval; FEV1, forced expiratory volume in 1 second; LS, least squares; q12h, every 12 hours; TEAE, treatment-emergent adverse event; ULN, upper limit of normal.

STUDY DESIGN
Trial 4 | Double-Blind, Placebo-Controlled Efficacy and Safety Study
Trial 4 was a 24-week, double-blind, placebo-controlled study assessing the efficacy and safety of ORKAMBI in patients age 6 through 11 years with CF who were homozygous for the F508del-CFTR mutation, were clinically stable, with an LCI2.5 score of ≥7.5, and a percent predicted FEV1 of ≥70%. Patients received either ORKAMBI tablets (lumacaftor 200 mg/ivacaftor 250 mg q12h) or placebo with fat-containing food and continued to take their prescribed CF therapies. Primary endpoint was an absolute change in LCI2.5 from baseline through Week 24.1,3
Additional Disclosure
- The Trial 4 efficacy results are not included in the approved full Prescribing Information, and the FDA did not consider either Trial 3 or Trial 4 study in approving ORKAMBI2
Safety Profile in Trial 4 Was Similar to That Observed in Patients Age 12 Years and Older1
Discontinuations Due to Adverse Events1,3
- Trial 4: 3% (n=3) in the ORKAMBI group vs 2% (n=2) in the placebo group discontinued treatment due to adverse events. These events included
- One case of abnormal respiration and two cases of elevated transaminases in the ORKAMBI group
- Two cases of elevated transaminases in the placebo group
Serious Adverse Reactions3
- Trial 4: 13% (n=13) in the ORKAMBI group vs 11% (n=11) in the placebo group experienced serious adverse events. Of these,
- Two were treatment related for ORKAMBI (one drug interaction and one obstructive airway disorder)
- Three were treatment related for placebo (one distal intestinal obstruction disorder and two elevated aminotransferases)
Liver-Related Adverse Reactions1,3
Potentially Clinically Significant (PCS) Laboratory Tests
| Parameter PCS/Categorical Criteria | TRIAL 4: 24 Weeks n/N (%) |
|
|---|---|---|
| ALT or AST | ORKAMBI | Placebo |
| >8 x ULN | 1/103 (1) | 2/101 (2) |
| >5 x ULN | 5/103 (5) | 3/101 (3) |
| >3 x ULN | 13/103 (13) | 8/101 (8) |
| Discontinuation due to transaminase elevations |
2/103 (2) | 2/101 (2) |
- In Trial 4, no patients had an increase in total bilirubin levels >2 x ULN1,3
Respiratory Adverse Events3
| TRIAL 4: 24 Weeks n/N (%) |
||
|---|---|---|
| ORKAMBI | Placebo | |
| Total Incidencec | 19/103 (18) | 13/101 (13) |
| Dyspnea | 5/103 (5) | 5/101 (5) |
| Respiration abnormal | 6/103 (6) | 4/101 (4) |
| Wheezing | 5/103 (5) | 3/101 (3) |
| Asthma | 4/103 (4) | 1/101 (1) |
| Chest discomfort | 0/103 (0) | 1/101 (1) |
cSome patients had more than one respiratory event.
Treatment Emergent Adverse Events1,3
Treatment Emergent Adverse Events With Incidence >10% in Any Treatment Group
| TRIAL 4d: 24 Weeks | ||
|---|---|---|
| ORKAMBI n=103 |
Placebo n=101 |
|
| Patients with any adverse event n (%) |
98 (95) | 98 (97) |
| Cough | 46 (45) | 47 (47) |
| Infective pulmonary exacerbation of cystic fibrosis | 20 (19) | 18 (18) |
| Productive cough | 18 (17) | 6 (6) |
| Nasal congestion | 17 (17) | 8 (8) |
| Oropharyngeal pain | 15 (15) | 10 (10) |
| Pyrexia | 15 (15) | 20 (20) |
| Upper abdominal pain | 13 (13) | 7 (7) |
| Headache | 13 (13) | 9 (9) |
| Upper respiratory tract infection | 13 (13) | 10 (10) |
| Sputum increased | 11 (11) | 2 (2) |
| Abdominal pain | 10 (10) | 10 (10) |
| Nausea | 10 (10) | 9 (9) |
| Rhinorrhea | 10 (10) | 5 (5) |
| Vomiting | 10 (10) | 10 (10) |
| Fatigue | 9 (9) | 11 (11) |
dTrial 4 Safety Set.
SUMMARY OF RESULTS
Lung Function Through 24 Weeks
Trial 4 | Statistically Significant Improvement Through Week 24 vs Placebo3


Adapted from Ratjen F et al. Lancet Respir Med. 2017;5(7):557-567, with permission from Elsevier.
Improvements in lung function are represented by a negative change in LCI.
- Within-group LS mean absolute change in LCI2.5 from baseline through Week 24 was -1.0 (95% CI -1.3 to -0.8) for ORKAMBI vs 0.1 (95% CI -0.2 to 0.3) for placebo. Treatment difference vs placebo is -1.1 (95% CI -1.4 to -0.8; P<0.0001)3
- LCI is still considered an exploratory endpoint in clinical trials4
ALT, alanine aminotransaminase; AST, aspartate aminotransaminase; CI, confidence interval; FEV1, forced expiratory volume in 1 second; LCI, lung clearance index; LS, least squares; q12h, every 12 hours; TEAE, treatment emergent adverse event; ULN, upper limit of normal.
Important Safety Information
Warnings and Precautions
Use in Patients With Advanced Liver Disease
- Worsening of liver function, including hepatic encephalopathy, in patients with advanced liver disease has been reported. Liver function decompensation, including liver failure leading to death, has been reported in CF patients with pre-existing cirrhosis with portal hypertension while receiving ORKAMBI. Use ORKAMBI with caution in patients with advanced liver disease and only if the benefits are expected to outweigh the risks. If ORKAMBI is used in these patients, they should be closely monitored after the initiation of treatment and the dose should be reduced
Indication and Usage
ORKAMBI is a combination of lumacaftor and ivacaftor indicated for the treatment of cystic fibrosis (CF) in patients aged 1 year and older who are homozygous for the F508del mutation in the CFTR gene. If the patient’s genotype is unknown, an FDA-cleared CF mutation test should be used to detect the presence of the F508del mutation on both alleles of the CFTR gene.
Limitations of Use
The efficacy and safety of ORKAMBI have not been established in patients with CF other than those homozygous for the F508del mutation.
Liver-related Events
- Serious adverse reactions related to elevated transaminases have been reported in patients with CF receiving ORKAMBI. In some instances, these elevations have been associated with concomitant elevations in total serum bilirubin
- It is recommended that ALT, AST, and bilirubin be assessed prior to initiating ORKAMBI, every 3 months during the first year of treatment, and annually thereafter. For patients with a history of ALT, AST, or bilirubin elevations, more frequent monitoring should be considered. Patients who develop increased ALT, AST, or bilirubin should be closely monitored until the abnormalities resolve
- Dosing should be interrupted in patients with ALT or AST >5 x upper limit of normal (ULN) when not associated with elevated bilirubin. Dosing should also be interrupted in patients with ALT or AST elevations >3 x ULN when associated with bilirubin elevations >2 x ULN. Following resolution of transaminase elevations, consider the benefits and risks of resuming dosing
Hypersensitivity Reactions, Including Anaphylaxis
- Hypersensitivity reactions, including cases of angioedema and anaphylaxis, have been reported in the postmarketing setting. If signs or symptoms of serious hypersensitivity reactions develop during treatment, discontinue ORKAMBI and institute appropriate therapy. Consider the benefits and risks for the individual patient to determine whether to resume treatment with ORKAMBI
Respiratory Events
- Respiratory events (e.g., chest discomfort, dyspnea, and respiration abnormal) were observed more commonly in patients during initiation of ORKAMBI compared to those who received placebo. These events have led to drug discontinuation and can be serious, particularly in patients with advanced lung disease (percent predicted FEV1 (ppFEV1) <40). Clinical experience in patients with ppFEV1 <40 is limited, and additional monitoring of these patients is recommended during initiation of therapy
Effect on Blood Pressure
- Increased blood pressure has been observed in some patients treated with ORKAMBI. Blood pressure should be monitored periodically in all patients being treated with ORKAMBI
Drug Interactions
- Substrates of CYP3A
Lumacaftor is a strong inducer of CYP3A. Administration of ORKAMBI may decrease systemic exposure of medicinal products that are substrates of CYP3A, which may decrease therapeutic effect. Co-administration with sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic index is not recommended. ORKAMBI may substantially decrease hormonal contraceptive exposure, reducing their effectiveness and increasing the incidence of menstruation-associated adverse reactions, e.g., amenorrhea, dysmenorrhea, menorrhagia, menstrual irregular. Hormonal contraceptives, including oral, injectable, transdermal, and implantable, should not be relied upon as an effective method of contraception when co-administered with ORKAMBI - Strong CYP3A Inducers
Ivacaftor is a substrate of CYP3A4 and CYP3A5 isoenzymes. Use of ORKAMBI with strong CYP3A inducers, such as rifampin, significantly reduces ivacaftor exposure, which may reduce the therapeutic effectiveness of ORKAMBI. Therefore, co-administration with strong CYP3A inducers is not recommended
Cataracts
- Cases of non-congenital lens opacities have been reported in pediatric patients treated with ORKAMBI and ivacaftor, a component of ORKAMBI. Although other risk factors were present in some cases (such as corticosteroid use and exposure to radiation), a possible risk attributable to ivacaftor cannot be excluded. Baseline and follow-up ophthalmological examinations are recommended in pediatric patients initiating treatment with ORKAMBI
ADVERSE REACTIONS
Serious Adverse Reactions
- Serious adverse reactions, whether considered drug-related or not by the investigators, that occurred more frequently in patients treated with ORKAMBI included pneumonia, hemoptysis, cough, increased blood creatine phosphokinase, and transaminase elevations. These occurred in 1% or less of patients
Most Common Adverse Reactions
- The most common adverse reactions in patients age 12 years and older in Phase 3 trials (Trials 1 and 2) occurring in ≥5% of patients treated with ORKAMBI (N=369) vs placebo (N=370) and at a rate higher than placebo were dyspnea, nasopharyngitis, nausea, diarrhea, upper respiratory tract infection, fatigue, respiration abnormal, blood creatine phosphokinase increased, rash, flatulence, rhinorrhea, and influenza
- The safety profile in patients age 6 through 11 years from an open-label trial (Trial 3; N=58) and a placebo-controlled trial (Trial 4; patients treated with ORKAMBI, N=103 vs placebo, N=101) was similar to that observed in Trials 1 and 2. Additional common adverse reactions were reported in Trial 4, but were not reported in Trials 1 and 2. The adverse reactions in Trial 4 that occurred in ≥5% of patients treated with ORKAMBI with an incidence of ≥3% higher than placebo included: productive cough, nasal congestion, headache, abdominal pain upper, and sputum increased
- The safety profile in patients age 2 through 5 years from an open-label trial (Trial 6; N=60) was similar to that in patients aged 6 years and older. The safety profile in patients age 1 through 2 years from an open-label trial (Trial 7; N=46) was similar to that in patients aged 2 years and older
USE IN SPECIFIC POPULATIONS
Pediatric Use
- The safety and effectiveness of ORKAMBI in patients with CF younger than 1 year of age have not been established
Click here to access full Prescribing Information for ORKAMBI.
References:
1. ORKAMBI [prescribing information]. Boston, MA: Vertex Pharmaceuticals Incorporated; August 2023. 2. Milla CE, Ratjen F, Marigowda G, et al.; On behalf of the VX13-809-011 Part B Investigator Group. Lumacaftor/ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017;195(7):912-920. doi: 10.1164/rccm.201608-1754OC